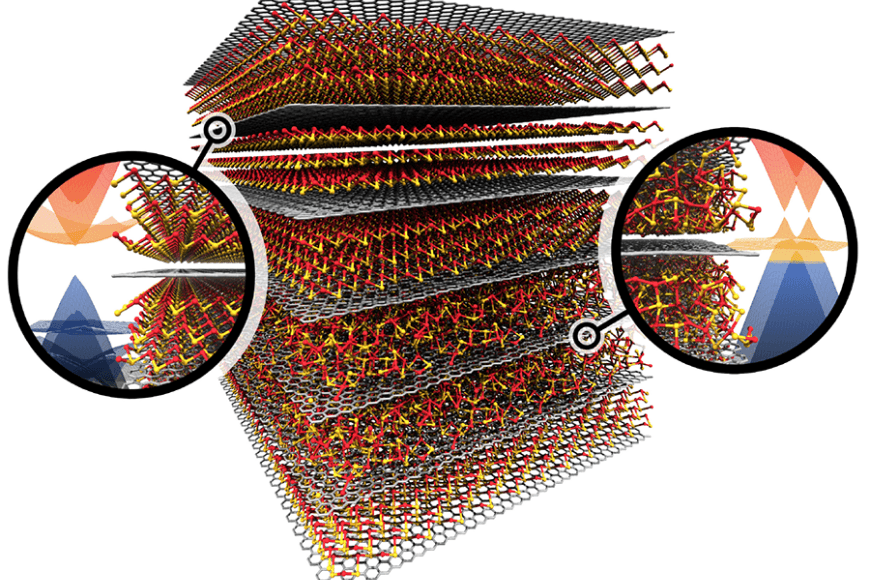
We perform simulations at the atomistic scale using both electronic structure calculations (DFT) and classical molecular mechanics (MM). The general objective our research is to study the detailed atomic structure of a system and its function. The group carries out method development for the Cluster Expansion formalism, Monte Carlo simulations, and tight-binding approaches. Furthermore, we develop simulation tools based on machine learning algorithms and link these with data mining.
Leader
Jaakko Akola
Professor, laskennallinen fysiikkaFaculty of Engineering and Natural Sciences
Tampere University
phone number0401981179
Hervanta Campus
Jaakko Akola